
Raquitismo dependiente de vitamina D tipo 1A por variante patogénica en CYP27B1: Reporte de caso
Autores/as
DOI:
https://doi.org/10.37980/im.journal.ggcl.es.20252759Palabras clave:
Raquitismo dependiente de vitamina D tipo 1A, proteína CYP27B1, hipocalcemia, fracturas patológicas, lactante, pruebas genéticasResumen
Caso clínico: Se describe el caso de un lactante masculino de un año de edad con antecedentes de desnutrición crónica y retraso global del desarrollo, quien ingresó al Hospital del Niño Dr. José Renán Esquivel con neumonía adquirida en la comunidad complicada con choque séptico. Hallazgos clínicos y radiológicos: Durante su hospitalización se documentaron alteraciones severas del metabolismo mineral, múltiples fracturas patológicas y hallazgos radiológicos compatibles con raquitismo severo. Hallazgos bioquímicos: Los estudios bioquímicos evidenciaron hipocalcemia persistente, elevación marcada de fosfatasa alcalina e hiperparatiroidismo secundario. Diagnóstico molecular: El estudio molecular mediante secuenciación de exoma completo identificó una variante patogénica homocigota en el gen CYP27B1 (NM_000785.4:c.602_611del; p.Val201AlafsTer31), confirmando el diagnóstico de raquitismo dependiente de vitamina D tipo 1A. Tratamiento y evolución: Se inició tratamiento con alfacalcidiol y suplementación de calcio, con respuesta clínica y bioquímica inicial favorable. Conclusión: Este caso resalta la importancia del reconocimiento oportuno de las formas genéticas de raquitismo y del uso de alternativas terapéuticas eficaces en contextos donde el calcitriol no está disponible.
Introducción
El raquitismo es un trastorno caracterizado por una mineralización defectuosa del cartílago de crecimiento, que ocurre exclusivamente en niños y se manifiesta clínicamente por deformidades óseas, retraso del crecimiento, hipotonía y, en casos severos, fracturas patológicas [1,2]. Aunque la causa más frecuente a nivel mundial continúa siendo la deficiencia nutricional de vitamina D, existen formas hereditarias de raquitismo que requieren un enfoque diagnóstico y terapéutico específico [3].
El raquitismo dependiente de vitamina D tipo 1A (VDDR1A; OMIM 264700) es una enfermedad rara de herencia autosómica recesiva causada por mutaciones en el gen CYP27B1, que codifica la enzima 25-hidroxivitamina D-1α-hidroxilasa renal [4]. Esta enzima es responsable de la conversión de 25-hidroxivitamina D en 1,25-dihidroxivitamina D (calcitriol), la forma biológicamente activa de la vitamina D [5]. La deficiencia de esta enzima conduce a hipocalcemia persistente, hiperparatiroidismo secundario, hipofosfatemia y elevación de fosfatasa alcalina, con compromiso óseo progresivo [6].
El tratamiento estándar del VDDR1A consiste en la administración de vitamina D activa (calcitriol) asociada a calcio. Sin embargo, en muchos países latinoamericanos el acceso al calcitriol es limitado, por lo que el alfacalcidiol representa una alternativa terapéutica eficaz y segura [7,8]. A continuación, se presenta un caso clínico representativo con manifestaciones severas y confirmación molecular.
Lactante masculino de un año de edad, procedente de área rural, con antecedentes de alimentación inadecuada y ausencia de suplementación previa con vitamina D u otros micronutrientes. Presentaba hipotonía generalizada y retraso global del desarrollo, sin deambulación independiente. Se documentó además desnutrición crónica. No se consignaron antecedentes familiares conocidos de raquitismo u otras enfermedades óseas hereditarias.
Ingresó por dificultad respiratoria progresiva, fiebre e irritabilidad. Al examen físico inicial se encontraba con hipoxemia, tiraje subcostal y mala perfusión periférica, evolucionando rápidamente a choque séptico de origen pulmonar.
Evolución hospitalaria
Durante la fase respiratoria aguda, se diagnosticó neumonía adquirida en la comunidad, requiriendo manejo en unidad de cuidados intensivos, oxígeno de alto flujo y soporte vasoactivo con norepinefrina. Durante la hospitalización, el hemocultivo fue positivo para Klebsiella pneumoniae BLEE; en consecuencia, se indicó tratamiento antibiótico dirigido. Recibió. Durante la evaluación integral se identificaron hallazgos óseos anormales, lo que motivó estudios radiológicos y metabólicos dirigidos.
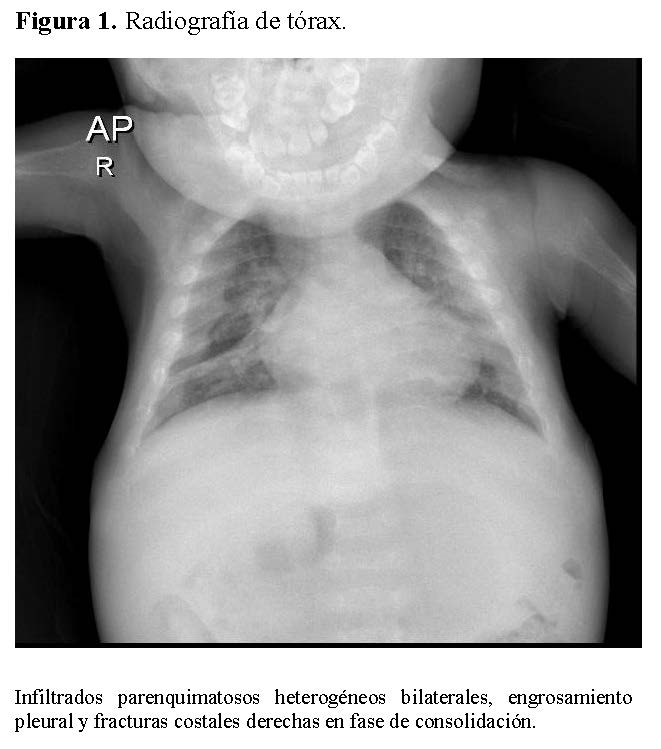
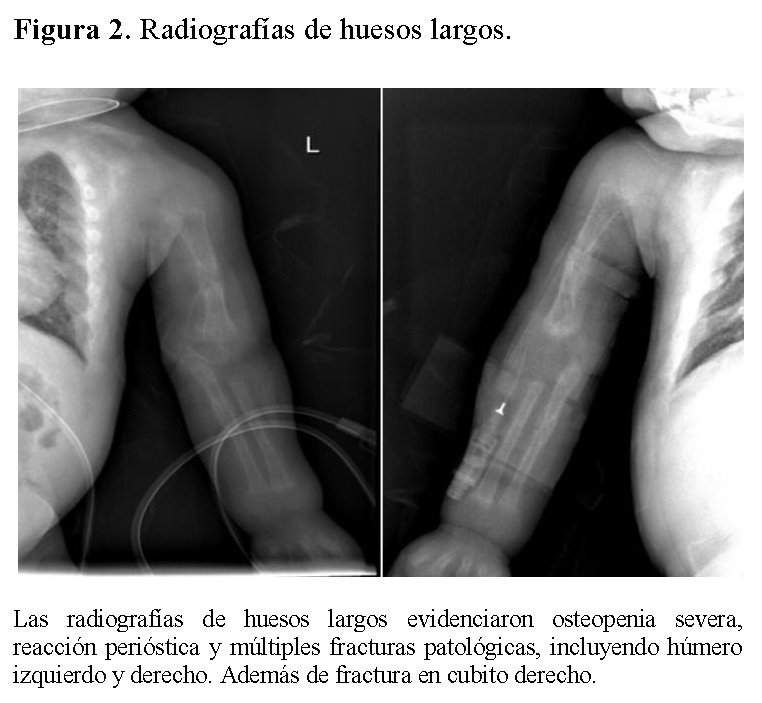
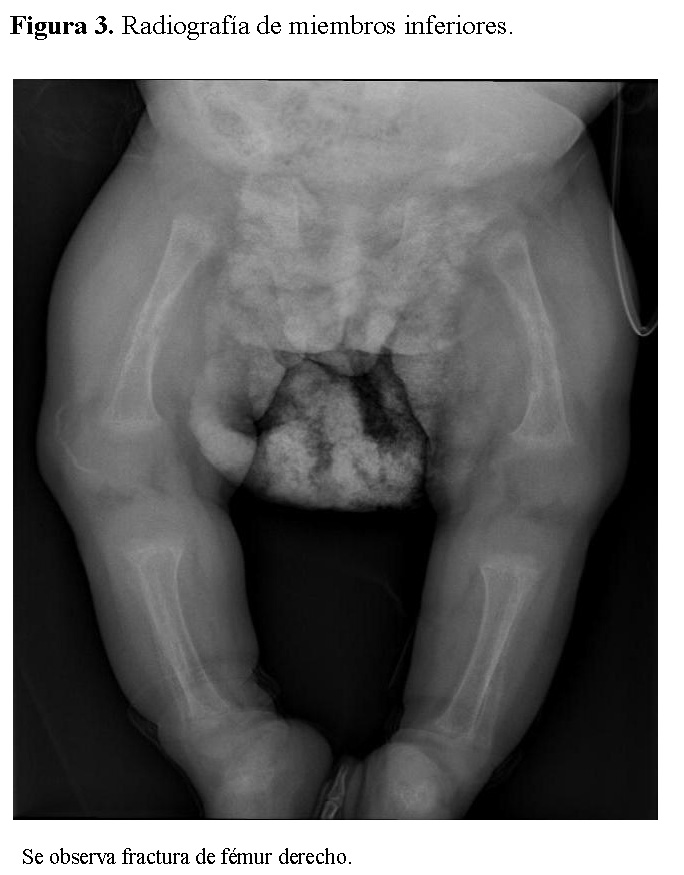
La radiografía de tórax mostró infiltrados parenquimatosos heterogéneos bilaterales, engrosamiento pleural y fracturas costales derechas en fase de consolidación (Figura 1). Las radiografías de huesos largos evidenciaron osteopenia severa, reacción perióstica y múltiples fracturas patológicas, incluyendo húmero derecho e izquierdo, cúbito derecho y fémur derecho (Figuras 2 y 3).
Resultados de laboratorio
Los estudios bioquímicos revelaron alteraciones severas del metabolismo mineral, con hipocalcemia persistente, hiperparatiroidismo secundario, elevación marcada de fosfatasa alcalina., niveles normales de 25-hidroxivitamina D y una marcada disminución de la 1,25-dihidroxivitamina D.
La combinación de un patrón de raquitismo calcipénico, caracterizado por hipocalcemia e hipofosfatemia, fosfatasa alcalina severamente elevada e hiperparatiroidismo secundario, junto con niveles normales de 25-hidroxivitamina D y una marcada disminución de la 1,25-dihidroxivitamina D, es altamente sugestiva de un defecto en la activación de la vitamina D.
El diagnóstico se sustentó en la integración de los hallazgos clínicos, radiológicos y bioquímicos, compatibles con un raquitismo de origen congénito. En este contexto, el perfil bioquímico observado fue compatible con deficiencia de 1-alfa-hidroxilasa, correspondiente al raquitismo dependiente de vitamina D tipo I, desde el punto de vista fisiopatológico y bioquímico.
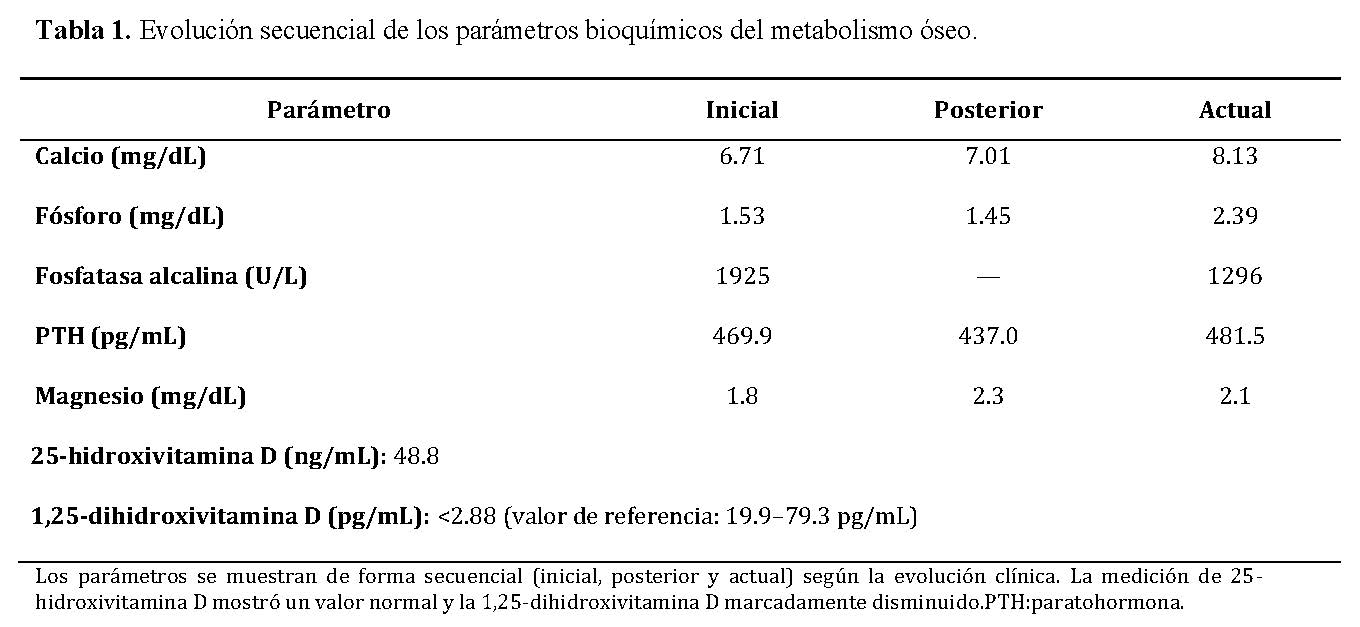
La evolución secuencial de los parámetros bioquímicos se resume en la Tabla 1, donde se evidencia hipocalcemia persistente, hiperparatiroidismo secundario marcado y descenso progresivo de la fosfatasa alcalina tras el inicio del tratamiento.
Estudio molecular
El análisis molecular se realizó mediante secuenciación de exoma completo, efectuada en un laboratorio externo acreditado (BGI Clinical Laboratories), identificándose una variante patogénica en homocigosis en el gen CYP27B1 (NM_000785.4:c.602_611del; p.Val201AlafsTer31). Esta variante corresponde a una deleción de 10 pares de bases que genera un deslizamiento del marco de lectura (frameshift) y un codón de terminación prematuro. Como consecuencia, se produce una proteína truncada y no funcional, con pérdida de la actividad de la 1α-hidroxilasa renal, lo que impide la síntesis de calcitriol y explica el fenotipo clínico observado.
Tratamiento y evolución
Se inició tratamiento sustitutivo con alfacalcidiol y gluconato de calcio ajustado a peso y respuesta bioquímica, junto con soporte nutricional intensivo y manejo ortopédico conservador. El paciente mostró mejoría progresiva del estado clínico, adecuada tolerancia oral, pero con dificultad para la estabilización de los parámetros bioquímicos, requiriendo hospitalización prolongada.
Discusión
El VDDR1A es una causa poco frecuente pero grave de raquitismo calciopénico, cuyo diagnóstico suele retrasarse debido a la similitud clínica con la deficiencia nutricional de vitamina D [4,6]. La deficiencia de la enzima 1-alfa-hidroxilasa renal impide la conversión de 25-hidroxivitamina D en 1,25-dihidroxivitamina D, lo que explica sus niveles marcadamente disminuidos pese a una 25-hidroxivitamina D normal. La reducción de vitamina D activa condiciona disminución de la absorción intestinal de calcio y fósforo, con desarrollo de hipocalcemia, hipofosfatemia e hiperparatiroidismo secundario, y un aumento del recambio óseo reflejado en la elevación severa de la fosfatasa alcalina, característico del raquitismo calcipénico. [4].
Las mutaciones tipo frameshift en CYP27B1 se asocian con pérdida completa de la función enzimática y fenotipos más severos, caracterizados por hipocalcemia profunda, hiperparatiroidismo secundario marcado y compromiso óseo temprano [9–10]. La variante se encuentra reportada en la base de datos ClinVar (Variation ID: 3575036), donde ha sido clasificada como probablemente patogénica. Sin embargo, considerando que se trata de una variante truncante con pérdida de función en un gen en el que este mecanismo es causa conocida de enfermedad, su detección en estado homocigoto y la clara concordancia con el fenotipo clínico del paciente, la variante fue clasificada como patogénica de acuerdo con los criterios del American College of Medical Genetics and Genomics (ACMG). La clasificación patogénica se sustentó principalmente en el criterio PVS1, complementado por los criterios PM2 y PM3. En este contexto, el uso de predictores bioinformáticos in silico no fue considerado necesario.
El diagnóstico molecular permitió confirmar la etiología, orientar el tratamiento específico que ya se había iniciado con el diagnostico bioquímico y brindar asesoramiento genético a la familia.
El calcitriol es el tratamiento de elección; sin embargo, múltiples estudios han demostrado que el alfacalcidiol es una alternativa eficaz cuando el calcitriol no está disponible, ya que solo requiere hidroxilación hepática para su activación [7,8,11]. En países latinoamericanos, donde la disponibilidad de calcitriol puede ser limitada, el alfacalcidiol representa una opción terapéutica segura y costo-efectiva, siempre que se realice un seguimiento estrecho de calcio y fósforo séricos [8,12].
Abreviaciones
· VDDR1A: Raquitismo dependiente de vitamina D tipo 1A; · PTH: Hormona paratiroidea; FA: Fosfatasa alcalina; 25-OH vitamina D: 25-hidroxivitamina D; 1,25-(OH) ₂ vitamina D: 1,25-dihidroxivitamina D (calcitriol); UTI: Unidad de Terapia Intensiva; BLEE: Beta-lactamasa de espectro extendido; OMIM: Online Mendelian Inheritance in Man;NAC: Neumonía adquirida en la comunidad.
Sobre los autores
KC: médica residente de genética clínica; OS: pediatra genetista clínico; KS: pediatra tratante; HL: endocrinóloga pediatra.
Agradecimientos
Se agradece al Dr. Jorge D. Méndez por su asesoramiento en la publicación de este manuscrito como proferor del Programa de Residencias en Génetica Clínica del Hospital del Niño, Panamá.
Referencias
[1] Thacher TD, Fischer PR. Vitamin D–deficiency rickets. N Engl J Med. 2011;364(3):248–254.
[2] Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357(3):266–281.
[3] Carpenter TO, Shaw NJ, Portale AA, Ward LM, Abrams SA, Pettifor JM. Rickets. Nat Rev Dis Primers. 2017;3:17101.
[4] Wang JT, Lin CJ, Burridge SM, Fu GK, Labuda M, Portale AA, Miller WL. Genetics of vitamin D 1α-hydroxylase deficiency in 17 families. Am J Hum Genet. 1998;63(6):1694–1702. doi:10.1086/302156.
[5] Christakos S, Dhawan P, Verstuyf A, Verlinden L, Carmeliet G. Vitamin D: metabolism, molecular mechanism of action, and pleiotropic effects. Physiol Rev. 2016;96(1):365-408. doi:10.1152/physrev.00014.2015.
[6] Fraser DR. Vitamin D–dependent rickets. Endocrinol Metab Clin North Am. 1989;18(3):637–648.
[7] Baroncelli GI, Bereket A, El Kholy M, Audì L, Cesur Y, Ozkan B, et al. Rickets in the Middle East: role of environment and genetic predisposition. J Endocrinol Invest. 2018;41(8):873–885.
[8] Pettifor JM. Calcium and vitamin D metabolism in children. Pediatr Nephrol. 2010;25(9):1839–1848.
[9] Demir K, Kattan WE, Zou M, Durmaz E, BinEssa H, Nalbantoğlu Ö, et al. Novel CYP27B1 gene mutations in patients with vitamin D-dependent rickets type 1A. PLoS One. 2015;10(6):e012.
[10] Tebben PJ, Milliner DS, Horst RL, Harris PC, Singh RJ, Wu Y, et al. Hypercalcemia, hypercalciuria, and nephrocalcinosis in CYP24A1 deficiency. Endocrinol Metab Clin North Am. 2017;46(4):875–893.
[11] Uday S, Högler W. Nutritional rickets and osteomalacia in the twenty-first century: revised concepts, public health, and prevention strategies. Curr Osteoporos Rep. 2017;15(4):293–302. doi:10.1007/s11914-017-0383-y.
[12] Kitanaka S, Takeyama K, Murayama A, Kato S. Molecular basis of vitamin D-dependent rickets type I. Bone. 2001;27(3):369–374.
